살면서 몇번이나 하게될지 모를, 그래서 까먹기 쉬운 프라이머 디자인 하는 법을 이참에 정리해보자. 특히 이번 포스트에서 정리할 내용은 종 특이적 (species-specific) 프라이머를 디자인하는 방법이다. 전체적인 workflow는 아래 논문을 참고하여 진행했다.
내가 최종적으로 디자인한 단계는 아래와 같이 총 5단계이다.

0. 데이터 모으기
우선 뭐라도 있어야 프라이머를 찾을게 아닌가! 내 목표는 종 특이적 프라이머를 찾는 것이기 때문에 목표로 하는 종의 가능한 많은 genome과 gene을 사용할 수록 좋다. 사용할 레퍼런스의 퀄리티도 중요하기 때문에, NCBI RefSeq DB에서 complete-level genome만 다운받아서 사용했다.
complete-level genome의 개수가 너무 적을 경우,
chromosome-level genome 까지 사용해도 무방할 것 같다.
scaffold 개수가 너무 조각나있지만 않고, cds region 개수가 비슷할 수록 좋다.
포스트를 작성하며 타겟한 종은 Staphylococcus xylosus 이다. NCBI RefSeq DB로부터 총 14개의 complete-level genome을 다운받을 수 있다. (포스트 작성일 기준) *_cds_from_genomic.fna (coding region sequence를 모아놓은 파일)을 하나의 파일로 합치면 총 37,177개의 유전자 서열을 얻을 수 있다.
1. Core gene 찾기
프라이머의 조건1: 해당 종의 모든 strain이 가지고있는 서열
가지고있는 모든 reference strain에 존재하는 유전자, core gene, 을 찾기위해 self-BLAST search를 활용한다. (identity > 80%)
1
2
3
4
5
# BLAST DB 만들기
makeblastdb -in S.xylosus.cds.fna -input_type fasta -dbtype nucl -out S.xylosus.cds
# BLAST search
blastn -db S.xylosus.cds -query S.xylosus.cds.fna -out S.xylosus.self.m6 -perc_identity 80 -outfmt 6 -max_target_seqs 37177 -num_threads 10
self-BLAST search를 할 때, max_target_seqs 옵션을 신경써주는게 좋다.
stand-alone BLAST의 경우 500, Web-BLAST의 경우 100이 기본 값이다.
해당 개수를 넘어가는 hit은 report하지 않기 때문에, 의도치 않은 결과를 만들어낼 수 있다.
넉넉하게 query sequence 개수만큼 지정해주면 모든 hit을 볼 수 있다.
self-BLAST 결과를 기준으로, type-strain의 gene들 중 다른 모든 strain에 hit을 보인 gene을 가져온다. (S. xylosus의 type-strain: SMQ-121; GCF_000709415.1)
총 2,490 genes중 2,277 genes가 core gene으로 찾아졌다. (굉장히 많다…💦)
2. unique gene 찾기
프라이머의 조건2: 다른 종에는 없는 서열
위에서 찾은 core gene이 사실 species-level core gene이 아니라 genus-level core gene이거나 house keeping gene일 수 있다. 그렇기 때문에 우리가 목적으로 하는 종에만 있는 유전자를 가려내야한다. 우선은 같은 속 (genus) 내에 없는 유전자를 추려내는 작업부터 시작한다.
똑같이 NCBI RefSeq DB에서 Staphylococcus reference genome을 모두 다운받아서 BLAST DB를 구축하고 search 한다. 당연히 S. xylosus는 제외! (identity > 50%)
1
2
3
4
5
# BLAST DB 만들기; 전체 genome 사용
makeblastdb -in Staphylococcus.wo_xylosus.fna -input_type fasta -dbtype nucl -out Staphylococcus.wo_xylosus
# BLAST search
blastn -db Staphylococcus.wo_xylosus -query S.xylosus.core.fna -out S.xylosus.genus.m6 -perc_identity 50 -outfmt 6 -num_threads 10
이번에는 max_target_seq 옵션을 신경쓰지 않아도 괜찮다.
어차피 하나라도 hit을 보이면 삭제할 것이기 때문에
하나의 hit이라도 보이는 경우 다른 종에서 관찰될 수 있는 유전자이므로 앞으로의 분석에서 제외해주도록 한다. 대부분의 유전자가 이 단계에서 필터링되는데, 2,277개 중 2,234개 유전자가 걸러졌다. 최종적으로 43개 유전자만 남았다.
3. Specificity check
2번 단계에서 같은 genus (Staphylococcus)에 대해서만 유일성을 조사했으므로, 모든 박테리아에대해 유일성을 조사하는 단계 역시 필요하다. 이 단계를 꼼꼼히 수행해야 좋은 퀄리티의 프라이머를 얻을 수 있다.
나는 주로 아래와 같은 3단계의 검증을 통과시킨다. (e-value < 0.05)
- All RefSeq complete genomes (local-BLAST)
- rep_prok_rep_genomes (web-BLAST)
- nt (web-BLAST)
첫번째 단계는 로컬 머신에 모든 RefSeq 서열을 다운받아야 가능하기 때문에 넘어가도 괜찮다. 우리 연구실에서는 1년에 2번 새로 추가된 서열들을 다운받기 때문에 가능했다.
S. xylosus의 경우 굉—-장히 비슷한 S. pseudoxylosus가 존재한다… 🙃
complete-genome은 없기 때문에 2번째 단계에서 걸러지지 않았다. 해당 종을 허용할지 말지에 따라 최종 candidates의 개수가 좀 달라진다. (이러한 옵션은 프로젝트마다 유동적으로 선택해야한다.)
| S. pseudoxylosus | candidates genes |
|---|---|
| allowed | 4 |
| not allowed | 28 |
4. Primer design
Program: Geneious Prime
Geneious Prime은 유료프그램이다 🥲 (굉장히 비싸다) 일단 30일 trial을 제공해주니까 빠르게 프로젝트를 끝내는 것을 추천한다. 이후에 또 써야될 일이 있으면 다음 선택지중에서 하나를 선택하면 된다.
- Geneious Prime 구독 (연구비 등)
- 개인의 여러 이메일 돌려쓰기
- 연구실 후배 이메일 빌려쓰기
30일 trial의 경우 제한되는 기능들이 몇개 존재한다. 프라이머 디자인하는데는 크게 지장이 없긴 하지만, 여러 다른 기능들을 유용하게 쓰고싶다면 그냥 구독하기로하자.
여기서부터 노가다의 시작이다. 정신 바짝 차리자
candidates genes중에 원하는 유전자를 고르고, 다른 모든 strain에 동일한 유전자 서열을 모두 모아서 따로 파일로 저장한다. 예를들어, >lcl|NZ_CP008724.1_cds_WP_029378137.1_283 유전자를 선택했다고 하면, S.xylosus.self.m6에서 해당 유전자가 hit을 보인 다른 유전자들을 모두 가져오면 된다. 총 14개의 strain이 있었으니, 14개의 gene이 모일 것이다.
파일을 빨간 네모 영역에 드래그앤드롭 하면 간단하게 import 할 수 있다.
프라이머 디자인을 수행하기 전, multiple sequence alignment (MSA)를 수행해야한다. 업로드한 서열을 마우스 오른쪽 클릭하고 Align/Assemble > Multiple Align...을 클릭하면 Alignment창이 뜬다. Geneious Alignment, MUSCLE Alignment, Clustal Omega등 여러가지 알고리즘이 뜨는데 원하는걸 아무거나 선택해도 무방하다. 난 주로 Clustal Omega를 사용한다. (어차피 다 비슷한 서열들을 넣었기 때문에 어떤걸 써도 큰 차이가 나지 않는다.)
MSA가 수행되면 ... Alignment라는 새로운 레코드가 생성된 것을 볼 수 있다. 해당 레코드를 오른쪽 클릭한 후 Primers > Design New primers...를 클릭하면 Design New Primers창이 뜬다. 기본 세팅으로하고 OK버튼을 누르면 디자인된 프라이머들이 화면 아래에 표시된다.
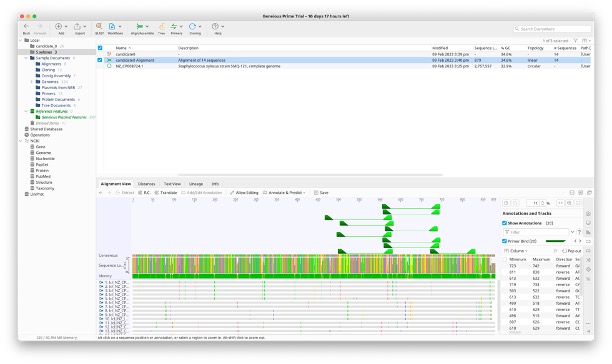
디자인된 프라이머를 아무거나 쓸 수 있는 것은 아니다. 다음과 같은 고려사항을 고려해서 프라이머 쌍을 선택하도록 하자.
- Primer 서열의 %GC (50.0 이상이 좋다)
- 두 Primer간의 Tm 차이 (1 미만이 좋다)
- Product size (200을 넘기는게 좋다; 100미만은 쓰지 말자)
- Primer에 해당하는 부분의 보존도 정도 (mismatch가 있는 곳은 피하는게 좋다)
mismatch가 전혀 없는 영역을 선택하는게 제일 좋겠지만, 힘들 경우 2개의 nucleotide가 등장하는 곳을 선택한 후 degrade 시키는 방법도 쓸만하다.
위에서 디자인한 프라이머쌍 중에 아래와 같은 한 쌍을 선택해봤다.
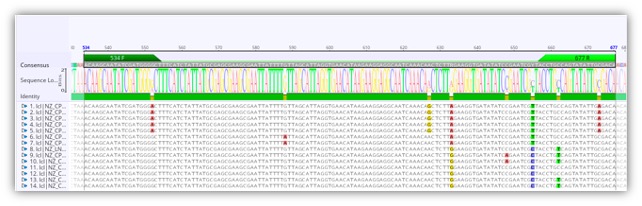
만족할만한 프라이머는 아니었다. 각 프라이머별로 1개씩 degrade를 시켜야하고, %GC도 50 미만이었다. product size도 144bp ㅠㅠ. 그나마 Tm 차이라도 적게나니까 다행인가….!!
이렇게 여러개를 도전해보고 적당한 프라이머쌍이 나올 때까지 위 과정을 반복해야한다!!! 중노동… 그러니까 적당히 석사생들에게 시키도록 하자 (읍읍)
추출할 프라이머 쌍을 선택한 뒤 Extract 버튼을 누르면 두 프라이머가 각각 레코드로 독립해나온다. 마지막으로 해당 프라이머쌍이 유전체에 딱 한번만 등장하는지를 검사해야한다.
목표로하는 종의 reference genome (type-strain의 genome을 추천한다)을 하나 업로드 해놓자. reference genome 레코드를 오른쪽 클릭한 뒤 Primers > Test with Saved Primers...를 클릭하면 Test with Saved Primers창이 뜬다. 방금 추출한 프라이머를 선택하고 다른 설정은 기본값으로 둔 뒤 OK 버튼을 누르자.

짜잔!! 전체 유전체에 대해서 한 번만 등장하는 것까지 확인했다!! 이제 마지막 단계만 남았다.
5. Double check
Program: Primer-BLAST
최종 관문만이 남았다. 앞에서 열심히 유전자를 골라가며 specificity를 검사했지만, 프라이머서열의 경우 20bp 정도로 짧다보니 다른 박테리아에 다시 map될 가능성이 여전히 존재한다.
NCBI에서 제공하는 Primer-BLAST를 이용해 최종적으로 specificity를 확인해보도록하자.
Primer-BLAST도 primer design tool이긴 하다. 이게 더 편하다면 이걸 써도 좋을 것 같다. 다만 전체 genome에 걸쳐서 한 번만 등장한다거나, degrade가 필요하다거나 등의 확인이 좀 어려울 수 있다.
Geneious Prime에도 Specificity-Checking 기능이 존재한다. 그러나 free 버전에서는 내가 직접 DB를 업로드 해줘야하고, Web-DB를 사용하고 싶으면 결제를 해야한다.
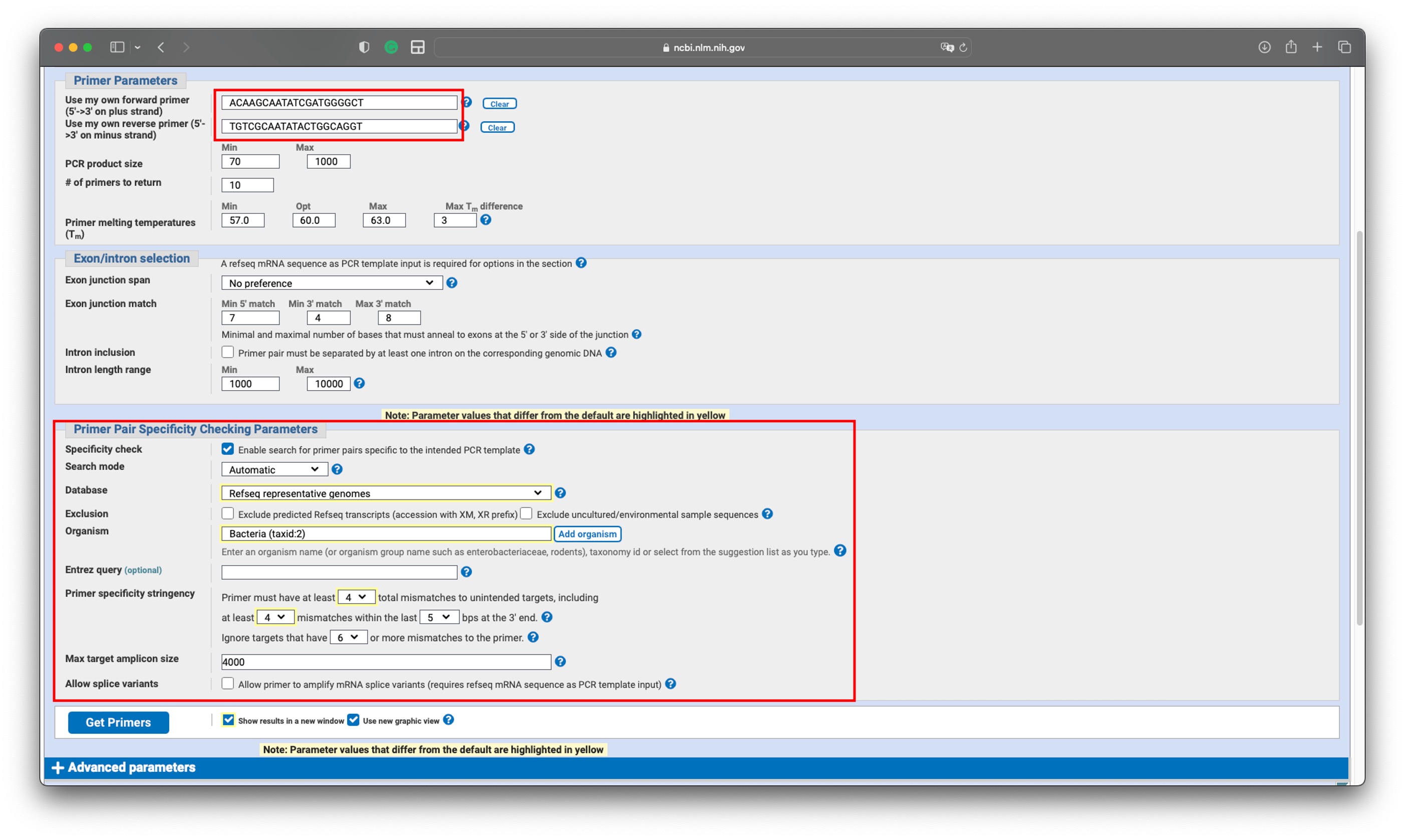
앞에서 디자인한 프라이머를 Primer Parameters 섹션에 넣어주고, Primer Pair Specificity Checking Parameters 섹션에서 DB를 Refseq representative genomes로 바꿔주자. Organism은 Bacteria (taxid:2)로 바꿔줘야 한다. 마지막으로 Primer specificity stringency에서 2라고 되어있는 값 2개를 둘 다 4로 변경해주자.
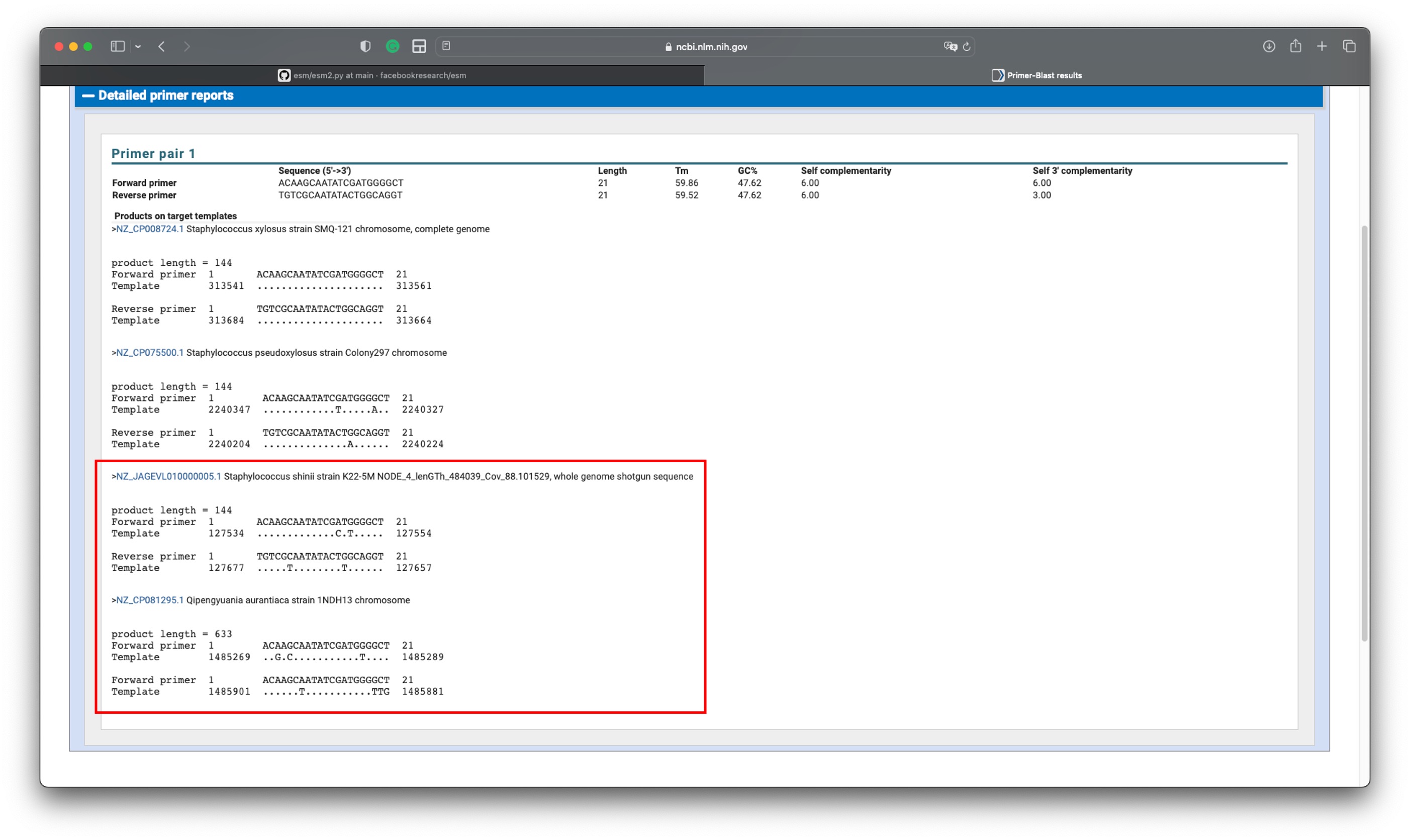
결과창에서 눈여겨 보아야 할 것은 딱 저 구간이다. 입력으로 넣어 준 Primer가 어떤 박테리아에 매핑되었는지를 알려주는 곳이다. 목적하는 종이 S. xylosus였으니 당연히 나와야 한다 (맨 윗줄). S. pseudoxylosus는 어느순간 일단 눈감아주고 있다.
절망적이게도, S. shinii와 Qipengyuania aurantiaca라는 종에 프라이머가 맵됐다… ㅠㅠ 일단 뒤에있는 어떻게 읽어야 할지도 모르겠는 녀석은 product length가 633이라고 나와있다. 이런 경우는 괜찮다!! 혹시 이녀석을 잡아내더라도, 훨씬 큰 사이즈의 DNA fragment가 생성될테니 전기영동에서 구분해 낼 수 있다. 하지만 저기 저 S. shinii를 보라🫣 product length도 절망적이게 똑같다. 이런 경우는 구분해 낼 수 없다.
그럼 이제 뭘해야할까?
당연히 다른 프라이머를 찾아 먼 길을 떠나야 한다👻🤑
그러니 난 이만 가보겠다. 아디오스